تصيب متلازمة هولت-أورام عظام اليدين والذراعين وقد تصيب أيضًا القلب. يكون لدى الأفراد المصابين بهذه المتلازمة عظم واحد على الأقل من عظام الرسغ لم يتشكل طبيعيًا. قد تتشكل عظام أخرى في اليدين والذراعين والكتف بصورة غير طبيعية. نرى الكثير من هذه التباينات التطورية عند الفحص بالأشعة السينية. يعاني معظم المصابون بهذه المتلازمة أيضًا اضطرابات قلبية تظهر نتيجة خلل خلقي في تكون القلب أو خلل في ضربات القلب.
تسبب التغيرات الجينية -تغيرات مرضية أو طفرات- في الجين (TBX5) هذه المتلازمة. يكون نمط الوراثة وراثةً جسديةً سائدة. يميل الطبيب إلى تشخيص هذه المتلازمة عندما تُلاحَظ الأعراض على المصاب، ويؤكَّد التشخيص بإجراء فحص بالأشعة السينية لليدين أو الرسغ أو الذراعين أو إجراء مخطط صدى القلب أو فحص جيني. تتضمن خيارات العلاج الخضوع للعمليات لإصلاح التشوهات العظمية ولعلاج المشكلات القلبية، إضافةً إلى جلسات العلاج الطبيعي.
الأعراض
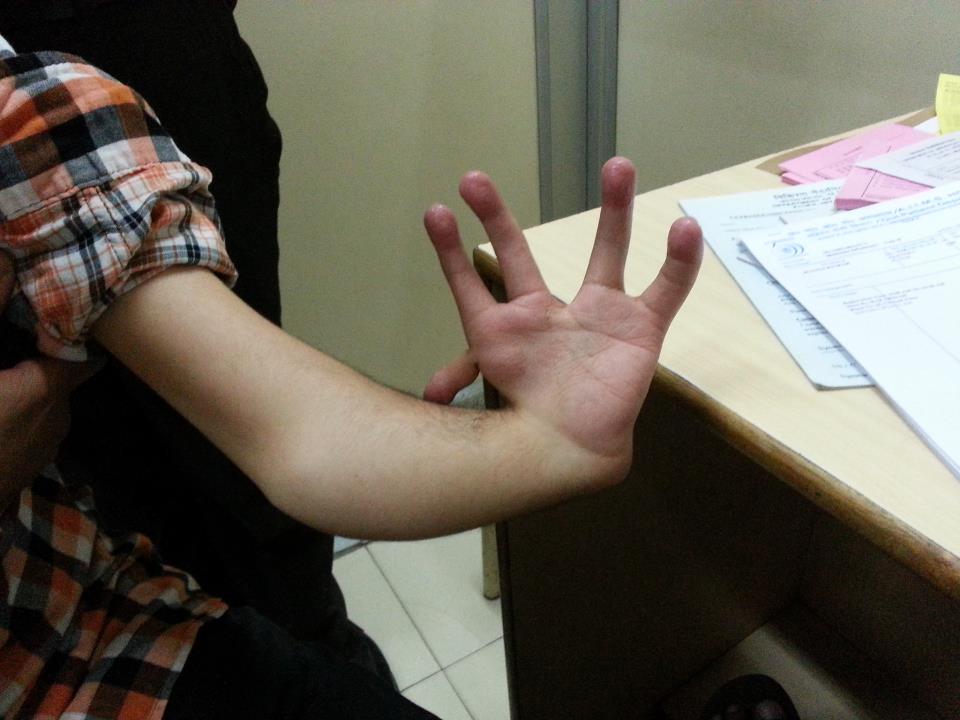
تتضمن الأعراض عيوب خلقية في اليدين والرسغ والذراعين والقلب. يكون لدى المصابين بهذه المتلازمة عظم واحد على الأقل من العظام الرسغية التي لم تتشكل طبيعيًا. تمتد التشوهات الشكلية المحتملة إلى عظام الطرف العلوي بأكمله، فقد يولد المصابون دون إصبع إبهام، أو بإبهام طويل يبدو مثل بقية الأصابع، أو باختفاء جزئي أو كامل لعظام الساعد والذراع، أو مشكلات في شكل عظم الترقوة أو لوح الكتف. قد يعجز المصابون عن بسط أذرعتهم تمامًا أو تدويرها. قد لا تظهر الشذوذات في بعض حالات الإصابة إلا بالفحص بالأشعة السينية.
يعاني نحو 75% من المصابين بمتلازمة هولت-أورام مشكلاتٍ قلبية، وتُعد الثقوب في الجدران القلبية التي تفصل بين الحجرات القلبية الأربعة أكثرها شيوعًا. يُعرف هذا الخلل باسم عيوب الحاجز الأذيني أو عيوب الحاجز البطيني اعتمادًا على مكان توضع الثقبة في الجدران. سُجلَت حالات الإصابة بقناة شريانية سالكة أيضًا. قد لا تسبب الاضطرابات القلبية الناجمة عن متلازمة هولت-أورام أي أعراض، وقد تسبب صعوبة في التنفس والإنهاك السريع -التعب- وارتفاع ضغط الدم في الشرايين الرئوية -فرط الضغط الرئوي- وصغر الحجم -قصور في النمو. قد تكون الاضطرابات القلبية مهددة للحياة في بعض الحالات.
يصاب بعض الأشخاص بمرض الناقلية القلبية، وهو اضطراب قدرة نظام التوصيل الكهربائي على التحكم في انتظام الضربات القلبية. قد يقود هذا المرض إلى بطء النظم القلبي -بطء القلب- أو إلى تقلصات قلبية سريعة وغير منتظمة -رجفان قلبي. تبدأ المشكلات الصحية المتعلقة بالناقلية القلبية بالظهور عند تقدم المصاب في العمر.
تتشابه أعراض متلازمة هولت-أورام مع متلازمة أخرى تسمى متلازمة دوان-راديال راي. تنتج المتلازمتان عن تغيرات جينية -تغيرات مرضية أو طفرات- في جينات متعددة. تترافق متلازمة هولت-أورام بمجال واسع من الأعراض والعلامات، حتى ضمن العائلة الواحدة، ما يعرف بـ «التباين التعبيري».
من الأعراض المسجلة:
- أعراض تظهر لدى 80-99% من المصابين: تشكل الترقوة الشاذ، تيبس المفاصل، يد مخلبية.
- أعراض تظهر لدى 30-79% من المصابين: شذوذات في عظام السنع -عظام اليد الطويلة-، غياب الإبهام، نقص التنسج/عدم التنسج الكعبري، عيوب الحاجز الأذيني -وجود فتحة في الحاجز الفاصل بين حجرتي القلب العلويتين-، إحصار أذيني بطيني من الدرجة الأولى، تحدب العمود الفقري، رجفان أذيني انتيابي، جنف، إبهام ثلاثي السلاميات -إبهام شبيه بالأصابع-، عيوب الحاجز البطيني -وجود فتحة في الحاجز الفاصل بين حجرتي القلب السفليتين.
- أعراض تظهر لدى 5-29% من المصابين: تشكل الشريان الأبهري الشاذ، تشوه عظم العضد، تشوه في الأضلاع، عيب العود الوريدي الرئوي الشاذ، عيب القناة الأذينية البطينية، إبهام عريض، كتفين مائلين للأسفل -كتفين مدورين-، ارتفاق الأصابع، متلازمة القلب الأيسر ناقص التنسج -نقص تصنع القلب الأيسر-، قناة شريانية سالكة، صدر مقعر -قمعي-، ارتفاق الأصابع، التحام عظمي الزند والكعبرة -التحام عظمي الساعد-، تشوه سبرينغل -ارتفاع الكتف الخلقي.
- أعراض تظهر لدى 1-4% من المصابين: غياب عظم الكعبرة -غياب عظم الساعد الكبير الخارجي-، عدم تنسج العضلة الصدرية الكبيرة، عدم تنسج عظم الزند، انحراف الأصابع، نقص تنسج عظم الكعبرة -نقص تنسج عظم الساعد الكبير الخارجي-، نقص تنسج عظم الزند -نقص تنسج عظم الساعد الكبير الداخلي-، محدودية القدرة على بسط المرفق، عيب الحاجز الأذيني الثانوي، قصر الترقوة، قصر الأصابع، قصر العضد -قصر العظم الطويل في الذراع-، صغر الرانفة، ارتفاق الأصابع -أصابع يدين وقدمين ذات أوتار كأقدام الأوز.
- أعراض غير محددة النسبة: تشكل الفقرات الشاذ، شذوذات العظام الرسغية، وراثة جسدية سائدة، تضاعف جزئي في عظام الإبهام، جنف صدري.
الأسباب
تنجم متلازمة هولت-أورام عن التغيرات -تغيرات مرضية أو طفرات- في جين (TBX5). يوفر هذا الجين المعلومات اللازمة لصنع البروتين المشارك في تطور القلب والأعضاء العلوية قبل الولادة. تكمن أهمية هذا الجين في توجيه القلب نحو الانقسام إلى أربع حجرات والتحكم بطريقة تشكل عظام الذراعين والرسغ واليدين. يتطور القلب والأطراف القلبية بصورة شاذة عندما يضطرب عمل هذا الجين، وهذا هو سبب ظهور أعراض متلازمة هولت-أورام.
وُجِدت فئة من المصابين بهذه المتلازمة الذين لا تظهر لديهم تبدلات مرضية في جين (TBX5). يبقى سبب المرض مجهولًا لدى هؤلاء.
الوراثة
ينتقل هذا المرض بوراثة جسدية سائدة. يكون لجين (TBX5) نسختين. تعني الوراثة الجسدية السائدة أن امتلاك نسخة واحدة متغيرة فحسب من جين (TBX5) كافٍ لظهور المرض والأعراض تباعًا. تأتي نسخة واحدة من هذا الجين من الأم والأخرى من الأب. عندما يُرزق المصاب بمتلازمة هولت-أورام بأطفال، تكون نسب الوراثة كالآتي:
- يوجد احتمال 50% أن يرث الطفل النسخة المتغيرة من جين (TBX5)، ما يعني إصابته بالمرض.
- يوجد احتمال 50% أن يرث النسخة السليمة، فلا يُصاب بالمرض.
يحدث التغير الجيني -التغير المرضي أو الطفرات- في جين (TBX5) لأول مرة عند الأطفال في 85% من حالات الإصابة بهولت-أورام من دون أن يكون هذا التغير موروثًا من أحد الأبوين. عندما يطرأ تغير مرضي لأول مرة يُسمى طفرة دي نوفو.
قد يرث الشخص المصاب بمتلازمة هولت-أورام في بعض الحالات النسخة المتغيرة من الجين من أحد الأبوين الحاملين لنسخة واحدة متغيرة، ولكن من دون علمهم بإصابتهم. يحدث هذا عندما تسبب الإصابة تغيرات طفيفة في عظام الرسغ ولا تسبب أي تغيرات أخرى في العظام الأخرى أو في القلب. تختلف أعراض الإصابة بمتلازمة هولت-أورام، حتى ضمن العائلة الواحدة، وهذا ما يعرف بالتباين التعبيري. لهذا السبب يصعب علينا توقع الأعراض وشدتها لدى الأطفال المستقبليين الحاملين لجين (TBX5) المتغيّر.
التشخيص
يتولد الشك بالإصابة بمتلازمة هولت-أورام عند ملاحظة تغيرات في تشكل عظام الرسغ وعظام الطرف العلوي الأخرى. يثبت التشخيص عند التأكد من وجود تغيرات محددة في العظام إضافةً إلى تاريخ مرضي لدى المصاب أو لدى عائلته من الإصابة بعيوب الحاجز الأذيني أو عيوب الحاجز البطيني أو مرض الناقلية القلبية. يطلب الطبيب للتحقق من التشخيص إجراء فحص بالأشعة السينية لليدين والرسغ والذراعين، وفحص لبنية القلب -مخطط صدى القلب- وفحص للنظم الكهربائي للقلب -مخطط كهربية القلب. قد يلجأ الطبيب إلى تأكيد التشخيص بإجراء فحص جيني للجين (TBX5).
العلاج
يتطلب العلاج طاقمًا من المختصين في طب الأطفال والجراحة والقلب وجراحة تقويم العظام والوراثة، ويتحدد هذا الطاقم بناءً على شدة الأعراض والمشكلات العظمية والقلبية الناتجة. قد نلجأ في علاج عظم الرسغ وعظام الطرف العلوي الأخرى إلى الجراحة الترميمية أو الإنشائية وإلى استخدام أطراف بديلة صناعية وإلى معالجة فيزيائية أو مهنية. تهدف المعالجة إلى مساعدة المصابين على التحكم بأطرافهم العلوية قدر المستطاع. تكمن الاستفادة العظمى من العلاج في حال البدء به فور التشخيص. قد لا تسبب التغيرات في بنية العظام مشكلات ولا تحتاج إلى علاج، فتبقى هذه التغيرات غير ملحوظة حتى تُكتشَف بالصدفة في أثناء إجراء فحص بالأشعة السينية لسبب آخر.
قد لا يحتاج المصابون بالاضطرابات القلبية المعتدلة أو بمرض الناقلية القلبية إلى علاج. يعالج مرض الناقلية القلبية في حالات أخرى بالأدوية المضادة لاضطراب النظم أو بزرع ناظم خطى للحفاظ على معدل منتظم لضربات القلب. قد تعالج الشذوذات القلبية الأخرى بالجراحة، ويعتمد الإجراء الجراحي على موضع الخلل وشدته.
يرتفع خطر الإصابة بالعدوى البكتيرية والتهاب بطانة القلب في الحجرات والصمامات القلبية -التهاب الشغاف- عند المصابين بالاضطرابات القلبية. قد يصف الطبيب المضادات الحيوية قبل إجراء الجراحة للتقليل من خطر العدوى.
مسار تطور المرض
يختلف مسار تطور المرض لدى المصابين بمتلازمة هولت-أورام بناءً على شدة الاضطرابات القلبية. لا يمتلك بعض المصابين أي مشكلات قلبية، وقد يمتلك بعضهم الآخر اضطرابات خفيفة الشدة تتطلب مراقبة دورية بواسطة اختصاصي الأمراض القلبية. قد تكون الاضطرابات القلبية في حالات أخرى شديدة أو مهددة للحياة.
قد تحدّ التغيرات في بنية عظام الرسغ واليدين والذراعين والكتف المترافقة مع متلازمة هولت-أورام قدرة المصاب الجسدية على أداء مهامه اليومية. قد تؤثر هذه التغيرات في التواصل الاجتماعي، وخصوصًا عند الأطفال الذين يمتلكون ذراعين ويدين ذات مظهر مختلف بوضوح عن بقية الأطفال.
اقرأ أيضًا:
متلازمة لازاروس: حين يعود الميت إلى الحياة!
متلازمة ستروم: الأسباب والأعراض والتشخيص والعلاج
ترجمة: ربا كيال
تدقيق: لبنى حمزة
الكاتب
ربا كيال
